
A new general molecule aligner in stko
Align two stk molecules
This is a short one because I have been busy (news to come!) but the key issue I aimed to show how to solve in this tutorial and post is the alignment of large, complex stk molecules without the constraints that the molecules must be the same or have the same atom orderings. Software like arbalign have solved this issue for small molecules, but I struggled to get the approach to work for large cage molecules.
My solution was to use SpinDry to iteratively find the optimal position of a host (the molecule to align to) and guest (the molecule to align) based on some potential. SpinDry has had a few new features recently and there are more to come but the main one is the ability to write custom potential functions through the spd.Potential classes. Using this, I wrote a potential function based on the distance between matched atom pairs. A matched atom pair is the pair of atoms of a defined pair of element types that are closest to each other. The element pair is defined by the user, but I tend to use the same element type (e.g. N-N or Pd-Pd). By iterating over multiple initial conformations and using SpinDry to generate low energy conformations based on this distance metric, the molecules can be aligned.
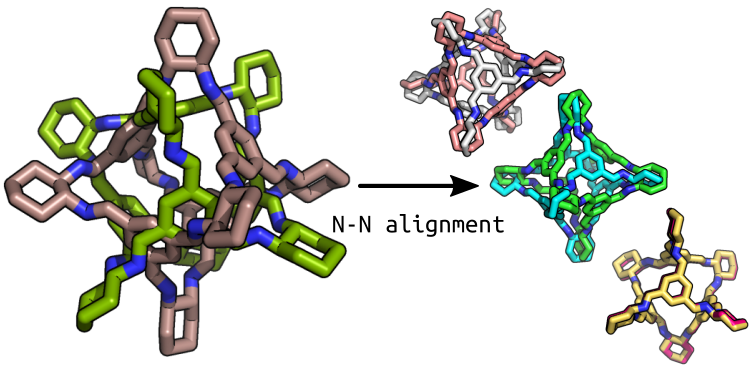
The Aligner Optimizer is implemented in stko and is now in the latest pip version. Additionally, two new RMSD calculators were added. The interface is as follows:
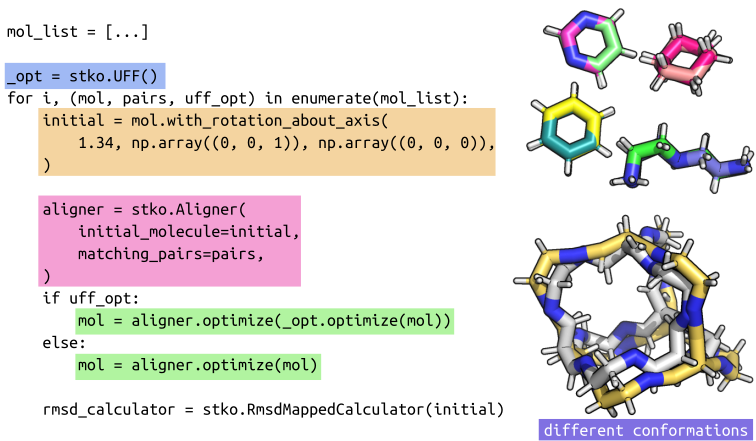
Fine-tuning can be necessary (the interface in stko will be improved in the future to make this easier), but in the tutorial, I show it is pretty general using the default settings. Indeed, the examples here and in the tutorial show it can be applied to molecules that are not the same. Ultimately though, there is likely a better option for aligning small molecules.
Examples are available in the stko examples directory and stk-examples.
Please, test it, use it, break it and send me feedback!